
And It's About Time There Was Some Support For Cushing's!
![]()
![]()
|
|
|
||||||||||||||||||
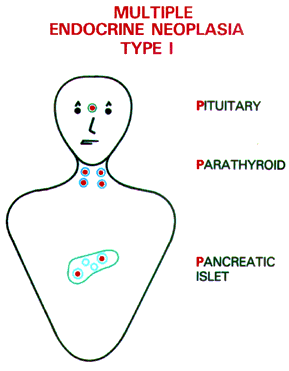
| Multiple endocrine neoplasia
type 1 (MEN1) is an inherited disorder that affects the endocrine glands. It
is sometimes called multiple endocrine adenomatosis or Wermer's syndrome,
after one of the first doctors to recognize it. MEN1 is quite rare,
occurring in about 3 to 20 persons out of 100,000. It affects both sexes
equally and shows no geographical, racial, or ethnic preferences.
Endocrine glands are different from other organs in the body because they release hormones into the bloodstream. Hormones are powerful chemicals that travel through the blood, controlling and instructing the functions of various organs. Normally, the hormones released by endocrine glands are carefully balanced to meet the body's needs. In patients with MEN1, sometimes more than one group of endocrine glands, such as the parathyroid, the pancreas, and the pituitary become overactive at the same time. Most people who develop overactivity of only one endocrine gland do not have MEN1. |
|||||||||||||||||||
|
How Does MEN1 Affect the Endocrine Glands? |
The Parathyroid Glands
The parathyroids are the endocrine glands earliest and most often affected by MEN1. The human body normally has four parathyroid glands, which are located close to the thyroid gland in the front of the neck. The parathyroids release into the bloodstream a chemical called parathyroid hormone, which helps maintain a normal supply of calcium in the blood, bones, and urine.
Nearly everyone who inherits a susceptibility to MEN1 (a "carrier") will develop overactive parathyroid glands (hyperparathyroidism) by age 50, but the disorder can often be detected before age 20. Hyperparathyroidism may cause no problems for many years or it may cause problems such as tiredness, weakness, muscle or bone pain, constipation, indigestion, kidney stones, or thinning of bones. Treatment of Hyperparathyroidism.
The pancreas gland, located behind the stomach, releases digestive juices into the intestines and releases key hormones into the bloodstream. Some hormones produced in the islet cells of the pancreas and their effects are:
Gastrin is another hormone that can be oversecreted in people with MEN1. The gastrin comes from one or more tumors in the pancreas and small intestine. Gastrin normally circulates in the blood, causing the stomach to secrete enough acid needed for digestion. If exposed to too much gastrin, the stomach releases excess acid, leading to the formation of severe ulcers in the stomach and small intestine. Too much gastrin can also cause serious diarrhea. About one in three patients with MEN1 has gastrin-releasing tumors, called gastrinomas. (The illness associated with these tumors is sometimes called Zollinger-Ellison syndrome.) The ulcers caused by gastrinomas are much more dangerous than typical stomach or intestinal ulcers; left untreated, they can cause rupture of the stomach or intestine and even death. Treatment of Gastrinomas.
The pituitary is a small gland inside the head, behind the bridge of the nose. Though small, it produces many important hormones that regulate basic body functions. The major pituitary hormones and their effects are:
The pituitary gland becomes overactive in about one of four persons with MEN1. This overactivity can usually be traced to a very small, benign tumor in the gland that releases too much prolactin, called a prolactinoma. High prolactin can cause excessive production of breast milk or it can interfere with fertility in women or with sex drive and fertility in men. Treatment of Prolactinomas. |
||||||||||||||||||
|
Rare Complications of MEN1 |
Occasionally, a person who has
MEN1 develops islet tumors of the pancreas that secrete high levels of
pancreatic hormones other than gastrin. Insulinomas, for example, produce
too much insulin, causing serious low blood sugar, or hypoglycemia.
Pancreatic tumors that secrete too much glucagon or somatostatin can cause
diabetes, and too much vasoactive intestinal peptide can cause diarrhea.
Other rare complications arise from pituitary tumors that release high amounts of ACTH, which in turn stimulates the adrenal glands to produce excess cortisol. Pituitary tumors that produce growth hormone cause excessive bone growth or disfigurement. Another rare complication is an endocrine tumor inside the chest or in the stomach, known as a carcinoid. In general, surgery is the mainstay of treatment for all of these rare types of tumors, except for gastric carcinoids which usually require no treatment. |
||||||||||||||||||
|
Are the Tumors Associated With MEN1 Cancerous? |
The overactive endocrine glands
associated with MEN1 may contain benign tumors, but usually they do not have
any signs of cancer. Benign tumors can disrupt normal function by releasing
hormones or by crowding nearby tissue. For example, a prolactinoma may
become quite large in someone with MEN1. As it grows, the tumor can press
against and damage the normal part of the pituitary gland or the nerves that
carry vision from the eyes. Sometimes impaired vision is the first sign of a
pituitary tumor in MEN1.
Another type of benign tumor often seen in people with MEN1 is a plum-sized, fatty tumor called a lipoma, which grows under the skin. Lipomas cause no health problems and can be removed by simple cosmetic surgery if desired. These tumors are also fairly common in the general population. Benign tumors do not spread to or invade other parts of the body. Cancer cells, by contrast, break away from the primary tumor and spread, or metastasize, to other parts of the body through the bloodstream or lymphatic system. The pancreatic islet cell tumors associated with MEN1 tend to be numerous and small, but most are benign and do not release active hormones into the blood. Eventually, about half of MEN1 cases will develop a cancerous pancreatic tumor. Treatment of Pancreatic Endocrine Cancer in MEN1. One approach is to "watch and wait," using medical, or nonsurgical treatments. According to this school of thought, pancreatic surgery has serious complications, so it should not be attempted unless it will cure a tumor that is secreting too much hormone. Another school advocates early surgery, perhaps when a tumor grows to a certain size, to remove pancreatic endocrine cancer in MEN1 (even if it does not over secrete a hormone) before it spreads and becomes dangerous. There is no clear evidence, however, that aggressive surgery to prevent pancreatic endocrine cancer from spreading actually leads to longer survival for patients with MEN1. Doctors agree that excessive release of certain hormones (such as gastrin) from pancreatic endocrine cancer in MEN1 needs to be treated, and medications are often effective in blocking the effects of these hormones. Some tumors, such as insulin-producing tumors of the pancreas, are usually benign and single and are curable by pancreatic surgery. Such surgery needs to be considered carefully in each patient's case. |
||||||||||||||||||
|
Is MEN1 the Same in Everyone? |
Although MEN1 tends to follow
certain patterns, it can affect a person's health in many different ways.
Not only do the features of MEN1 vary among members of the same family, but
some families with MEN1 tend to have a higher rate of prolactin-secreting
pituitary tumors and a much lower frequency of gastrin-secreting tumors.
In addition, the age at which MEN1 can begin to cause endocrine gland overfunction can differ strikingly from one family member to another. One person may have only mild hyperparathyroidism beginning at age 50, while a relative may develop complications from tumors of the parathyroid, pancreas, and pituitary by age 20. Sometimes a patient with MEN1 knows of no other case of MEN1 among relatives. The commonest explanations are that knowledge about the family is incomplete or that the patient carries a new MEN1 gene mutation. |
||||||||||||||||||
|
Can MEN1 Be Cured? |
There is no cure for MEN1
itself, but most of the health problems caused by MEN1 can be recognized at
an early stage and controlled or treated before they become serious
problems.
If you have been diagnosed with MEN1, it is important to get periodic checkups because MEN1 can affect different glands, and even after treatment, residual tissue can grow back. Careful monitoring enables your doctor to adjust your treatment as needed and to check for any new disturbances caused by MEN1. Most people with MEN1 will have long and productive lives. |
||||||||||||||||||
|
How Is MEN1 Detected? |
Each of us has millions of
genes in each of our cells, which determine how our cells and bodies
function. In people with MEN1, there is a mutation, or mistake, in one gene
of every cell. A carrier is a person who has the MEN1 gene mutation.
The MEN1 gene mutation is transmitted directly to a child from a
parent carrying the gene mutation.
The MEN1 gene was very recently identified. As of 2001, a small number of centers around the world have begun to offer MEN1 gene testing on a research or commercial basis. The likelihood of finding a mutation in an MEN1 family has varied from 60 percent to 94 percent depending on methods. When a mutation is found, further testing in other relatives can become much easier. Many relatives can be tested once and be found without the known MEN1 mutation in their family, and then they can be freed from uncertainty and from any further testing ever for MEN1. When a mutation is not found in a family or isolated case, it does not prove that no MEN1 mutation is present. Depending on the clinical and laboratory information, it may still be very likely that a mutation is present but undetected. In the meantime, though, screening of close relatives of persons with MEN1, who are at high risk, generally involves testing for hyperparathyroidism, the most common and usually the earliest sign of MEN1. Any doctor can screen for hyperparathyroidism by testing the blood for total calcium and sometimes one or two other substances such as ionized calcium and parathyroid hormone. An abnormal result indicates that the person probably has MEN1, but a normal finding in all cannot rule out the chance that he or she will develop hyperparathyroidism at a later time. Blood testing can usually show signs of early hyperparathyroidism many years before symptoms of hyperparathyroidism occur. |
||||||||||||||||||
|
What is the Role for Genetic Counseling with MEN1 Gene Testing? |
Genetic counseling, which
should accompany the gene testing, can assist family member(s) to address
how the test results affect them individually and as a family. In genetic
counseling, there can be a review and discussion of issues about the
psychosocial benefits and risks of the genetic testing results. Genetic
testing results can affect self-image, self-esteem, and individual and
family identity. In genetic counseling, issues related to how and with whom
genetic test results will be shared and their possible effect on important
matters such as health and life insurance coverage can be reviewed and
discussed. The times for these discussions can be when a family member is
deciding whether or not to go ahead with the gene testing and again later
when the gene testing results are available. The person, who provides the
genetic counseling to the family member(s), may be a professional from the
disciplines of genetics, nursing, or medicine. |
||||||||||||||||||
|
Who Should Consider MEN1 Screening? |
Who Should Consider MEN1 Screening by Gene Testing? Screening may be offered to persons with MEN1 or with features resembling it. Affected relatives of persons with MEN1 can be tested. Asymptomatic offspring, brothers, or sisters of a person with MEN1 were born with a 50 percent chance of having inherited the gene; they too can be offered gene testing. While gene testing for certain genes can be definitive at any age, it is usually not offered to children below age 18 unless the test outcome would have an important effect on their medical treatment. Since treatable tumors occasionally begin by age 5 in MEN1, gene testing and tumor surveillance can begin at age 5. Who Should Consider MEN1 Screening by Laboratory Tests? MEN1 screening by gene testing will be the most definitive test, when it is available. However, it is not yet widely available, and, when no gene mutation is found in a MEN1 family, then it may be necessary to rely upon laboratory tests for diagnosis. Hyperparathyroidism, most often the first sign of MEN1, can usually be detected by blood tests between the ages of 15 and 50. Periodic testing should begin around age 10 and be repeated every year. There is no age at which periodic testing should stop, since doctors cannot rule out the chance that a person has inherited the MEN1 gene mutation. However, a person with normal testing beyond age 50 is very unlikely to have inherited the MEN1 gene mutation. |
||||||||||||||||||
|
Why Screen for MEN1 Tumors? |
MEN1 is not an infectious or
contagious disease, nor is it caused by environmental factors. Because MEN1
is a genetic disorder inherited from one parent, and its transmission
pattern is well understood, family members at high risk for the disorder can
be easily identified.
Testing can detect the problems caused by MEN1 tumors many years before their later complications develop. Finding these tumors early enables your doctor to begin preventive treatment, reducing the chances that MEN1 will cause problems later. |
||||||||||||||||||
|
Should a Person Who Has MEN1 Avoid Having Children? |
A person who has MEN1 or who
has a positive MEN1 gene mutation may have a hard time deciding
whether to have a child. No one can make this decision for anyone else, but
some of the important facts can be summarized as follows:
|
||||||||||||||||||
|
Research in MEN1 |
The National Institute of
Diabetes and Digestive and Kidney Diseases (NIDDK) was established by
Congress in 1950 as part of the National Institutes of Health (NIH), whose
mission is to improve human health through biomedical research. The NIH is
the research arm of the Public Health Service under the U.S. Department of
Health and Human Services.
The NIDDK conducts and supports a variety of research in endocrine disorders, including MEN1. NIDDK and other NIH researchers isolated the MEN1 gene in 1997. Researchers have also shown that the MEN1 gene contributes to common endocrine tumors outside of the setting. |
||||||||||||||||||
|
Additional Information |
After reading this e-text, you
may think of questions that you would like answered. Some sources of
additional information are medical textbooks, physicians, nurses, and
genetic counselors.
The following articles about MEN1 can be found in medical libraries, some college and university libraries, and through interlibrary loan in most public libraries. Chandrasekharappa, S.C., Guru, S.C., Manickam, P., Olufemi, S., Collins, F.S. Emmert-Buck, M.R., Debelenko, L.V., Zhuang, Z., Lubensky, I.A., Liotta, L.A., Crabtree, J.S., Wang, Y., Roe, B.A., Weisemann, J., Boguski, M.S., Agarwal, S.K., Kester, M.B., Kim, Y.S., Heppner, C., Dong, Q., Spiegel, A.M., Burns, A.L., Marx, S.J., "Positional cloning of the gene for multiple endocrine neoplasia-type 1," Science 276:404-407, 1997. Marx SJ. Multiple endocrine neoplasia type 1. In: Metabolic Basis of Inherited Diseases, 8th Ed. ed. Scriver CS, et al. McGraw Hill, NY, 2001. pp 943-966. Schussheim DH, Skarulis MC, Agarwal SK, Simonds WF, Burns AL, Spiegel AM, Marx SJ. MEN1: New clinical and basic findings. Trends Endocrinol Metab 12: 173-178, 2001. |
||||||||||||||||||
|
Other Resources |
The following organizations
might also be able to assist with certain types of information:
Pituitary Network Association Office of Scientific and Health Information March of Dimes/Birth Defects Foundation Alliance of Genetic Support Groups This e-pub was written by Stephen J. Marx, M.D. and reviewed by Robert T. Jensen, M.D., both of the National Institute of Diabetes and Digestive and Kidney Diseases. It is based in part on the booklet, "Understanding Multiple Endocrine Neoplasia Type 1," published by the NIH Clinical Center's Communications Office.
|
||||||||||||||||||
|
|
NIH Publication No. 02-3048 June 2002 |
 In
MEN1, all four parathyroid glands tend to be overactive. They release too
much parathyroid hormone, leading to excess calcium in the blood. High blood
calcium, known as hypercalcemia, can exist for many years before it is found
by accident or by family screening. Unrecognized hypercalcemia can cause
excess calcium to spill into the urine, leading to kidney stones or kidney
damage.
In
MEN1, all four parathyroid glands tend to be overactive. They release too
much parathyroid hormone, leading to excess calcium in the blood. High blood
calcium, known as hypercalcemia, can exist for many years before it is found
by accident or by family screening. Unrecognized hypercalcemia can cause
excess calcium to spill into the urine, leading to kidney stones or kidney
damage.